|
La sclerosi laterale amiotrofica (SLA)
|
Cari ragazzi questo approfondimento nasce per rispondere ad una domanda di Linda Ficara della classe 3a C e ci permette di capire quanto sia complesso
lo studio di malattie terribili come la sclerosi laterale amiotrofica (SLA). Vi ricordate? Si tratta della malattia di Stephen Hawking.
La SLA è una malattia rara, colpisce 2-3 persone su 100000 all'anno. Attacca i motoneuroni, quei neuroni che muovono i muscoli volontari, in particolare i fasci di
neuroni laterali del midollo spinale, che collegano la corteccia cerebrale con le corna del midollo spinale. Queste cellule degenerano
fino a morire e il paziente affetto da SLA perde progressivamente
la capacità di muoversi. Sia i motoneuroni cervicali che spinali finiscono per degenerare e non inviano più segnali ai muscoli, che si atrofizzano e si
induriscono, diventando, appunto, sclerotici. La malattia insorge in età adulta
e in genere porta alla morte per paralisi respiratoria entro pochi anni dalla comparsa dei primi sintomi. In alcuni casi, come quello di Stephen Hawking, il decorso è più lento. Come per altre malattie
neurodegenerative, esistono due forme di SLA: una ereditaria (il 10 % dei casi) e una sporadica (il 90 % dei casi). Dal 1993 ad oggi sono stati
identificati 9 geni le cui mutazioni provocano il 68 % dei casi di SLA ereditaria e sono implicati anche nell'11 % dei casi di SLA sporadica.
Quali sono le funzioni di questi geni? E cosa accade quando sono mutati?
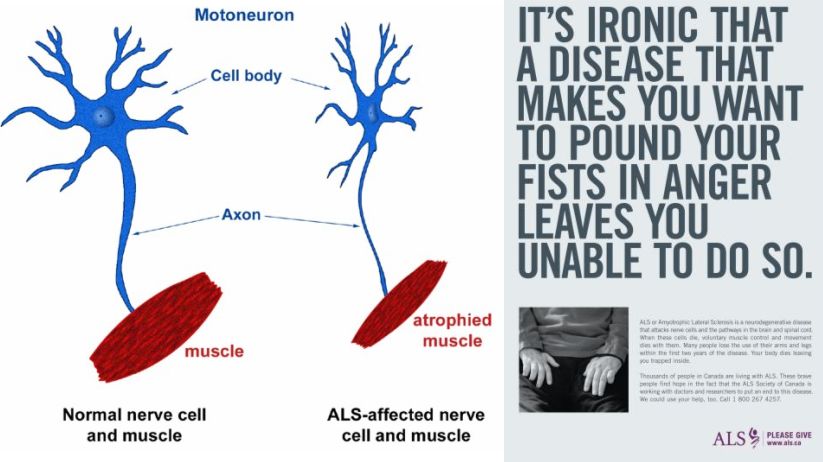 |
La sclerosi laterale amiotorfica porta alla graduale degenerazione e morte dei motoneuroni, cioè i neuroni che controllano i muscoli volontari.
(http://4.bp.blogspot.com/_WatmprlJuSk/TCJggCxHU4I/AAAAAAAAAPI/AkooAnasT6w/s1600/ALS1.jpg
http://files.coloribus.com/files/adsarchive/part_744/7449355/file/amyotrophic-lateral-sclerosis-charity-pound-your-fists-small-91385.jpg)
|
Consideriamo due esempi, i geni SOD1 e C9ORF72, che spiegano insieme circa il 50 % dei casi di SLA ereditaria e il 10 % di quelli sporadici.
Il gene SOD1 codifica
per la proteina superossido dismutasi, che ha il compito di distruggere ioni superossido nel nostro organismo. Questi ioni, noti come radicali liberi,
si formano, come prodotti di scarto, durante la respirazione cellulare nei mitocondri e vengono anche usati dal nostro sistema immunitario per uccidere i microorganismi
patogeni. Questi superossidi sono molto dannosi per le nostre cellule. Infatti, possono ossidare i lipidi delle membrane causando danni irreparabili.
Per questo motivo abbiamo il gene SOD1, che produce una proteina che detossifica i superossidi. Se il gene SOD1 è mutato, le cellule subiscono danni e la stessa
proteina superossido dismutasi mutata si accumula all'esterno delle cellule diventando tossica. Secondo alcuni studiosi è proprio questa attività tossica che
porterebbe alla degenerazione dei neuroni nella SLA. La mutazione è dominante, basta una sola copia del gene SOD1 mutato per sviluppare la SLA.
La mutazione nel gene C9ORF72 è molto particolare. La funzione stessa di questo gene non è ancora chiara agli scienziati. Nel gene non mutato, in una regione che
non codifica per gli aminoacidi, ma probabilmente coinvolta nella regolazione della sua espressione, ci sono circa 30 ripetizioni di sei nucleotidi, GGGGCC.
Nella forma mutata questa
ripetizione è presente centinaia di volte. Questa mutazione è di estremo interesse per vari motivi. Non solo è quella che spiega la maggior parte dei casi di SLA, ma è
anche associata alla sindrome FTD ("frontotemporal dementia"), una forma di demenza senile. Alcuni casi di SLA, sono complicati
dall'insorgenza di questa demenza e in entrambe le patologie è coinvolta questa ripetizione dell'esanucleotide GGGGCC. Queste strane ripetizioni di più basi sono coinvolte anche
in un'altra terribile malattia genetica neurodegenerativa, la corea di Huntington.
Altri geni sono già stati identificati e studiati. Molti di questi sono coinvolti nel funzionamento dell'RNA e della regolazione dell'espressione genica e gli scienziati stanno
cercando di capire i meccanismi molecolari che portano alla degenerazione dei neuroni nella SLA.
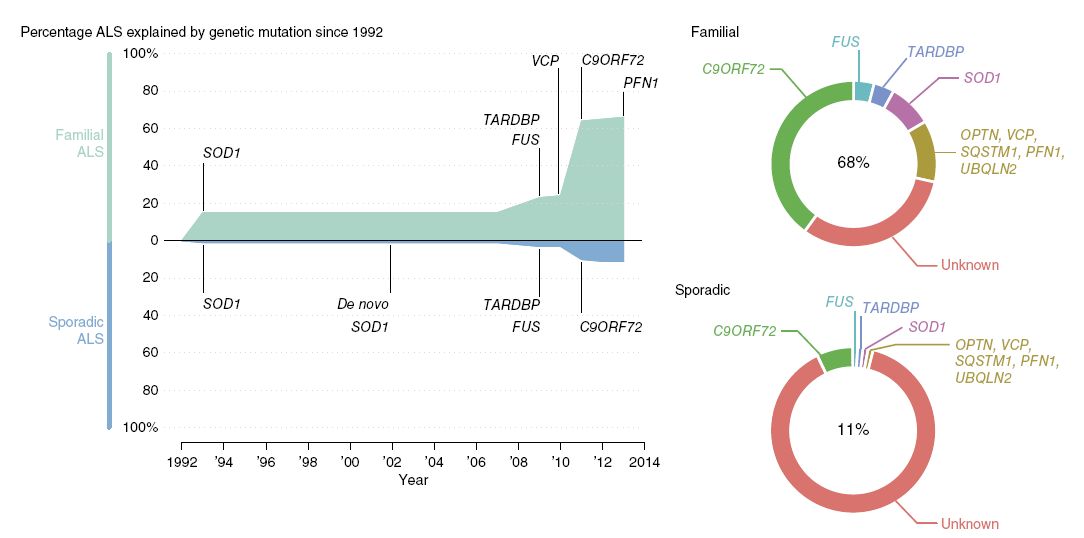 |
Il grafico riporta la percentuale di casi di SLA ereditaria e sporadica, causati dai vari geni mutati. Nel 68 % dei casi di SLA familiare si conoscono i geni
coinvolti.
Alcune di queste mutazioni si ritrovano anche nei pazienti con SLA non familiare (nell'11 % dei casi).
(Ripreso da Renton et al., 2014)
|
Ora che alcuni geni e le loro mutazioni responsabili della SLA sono stati identificati, cosa si fa? Sono stati creati diversi modelli animali. Per esempio il gene
SOD1 è stato mutato artificialmente in cavie da laboratorio, si è visto che compaiono sintomi simili a quelli della SLA umana e si sta studiando nel modello il decorso
della malattia e gli eventi molecolari che portano alla degenerazione dei neuroni. Perché? Perché solo se si riesce a comprendere la biochimica dei processi molecolari che
sono alla base della morte dei neuroni si può, poi, progettare un intervento genetico mirato ad evitare che ciò accada. Ed è proprio dalla studio dei modelli animali che si
è capito che la morte dei neuroni deriva da difetti nei processi che coinvolgono l'RNA, il riciclaggio delle proteine, l'iperattivazione dei neuroni o di
cellule infiammatorie della glia, cellule che formano il sistema nervoso insieme ai neuroni. Questi studi hanno aperto la strada sia a sperimentazioni in vivo
di farmaci genetici volti a bloccare la degenerazione dei neuroni sia all'identificazione di biomarcatori per una diagnosi precoce della SLA.
Alcuni ricercatori si sono, invece, concentrati sulla possibilità di usare cellule staminali pluripotenti indotte, derivate dai tessuti del malato di SLA.
I risultati, purtroppo, non sono ancora confortanti.
Non bisogna inoltre dimenticare che circa il 90 % dei casi di SLA è sporadico, cioè non ereditario e, sfortunatamente, nella maggior parte di questi casi, la causa della
malattia è ancora ignota. E' come se la SLA non fosse una singola malattia, ma un insieme di patologie diverse, che conducono tutte alla degenerazione e morte dei
motoneuroni.
No, non è per niente facile trovare una cura per una malattia così complessa.
Se volete, leggete la storia di Luca Coscioni, un coraggioso orvietano morto di SLA nel 2006.
Referenze
Renton A.E., Chiò A., Traynor B.J. (2014) State of play in amyotrophic lateral sclerosis genetics. Nature Neuroscience, 17 17-23.
Bento-Abreu a., Van Damme P., Van Den Bosch L., Robberecht W. (2010) The neurobiology of amyotrophic lateral sclerosis. European Journal of Neuroscience, 31: 2247–2265.
Bowerman M., Vincent T., Scamps F., Perrin FE., Camu W., Raoul C. (2013) Neuroimmunity dynamics and the development of therapeutic strategies for amyotrophic lateral
sclerosis. Frontiers in cellular neurosciences, 7: 1-10.
Manuela Casasoli (manuela_casasoli@yahoo.it)